Welcome to sourmash!¶
sourmash is a command-line tool and Python library for computing hash sketches from DNA sequences, comparing them to each other, and plotting the results. This allows you to estimate sequence similarity between even very large data sets quickly and accurately.
sourmash can be used to quickly search large databases of genomes for matches to query genomes and metagenomes; see our list of available databases.
sourmash also includes k-mer based taxonomic exploration and classification routines for genome and metagenome analysis. These routines can use the NCBI taxonomy but do not depend on it in any way.
We have several tutorials available! Start with Making signatures, comparing, and searching.
The paper Large-scale sequence comparisons with sourmash (Pierce et al., 2019) gives an overview of how sourmash works and what its major use cases are. Please also see the mash software and the mash paper (Ondov et al., 2016) for background information on how and why MinHash works.
Questions? Thoughts? Ask us on the sourmash issue tracker!
To use sourmash, you must be comfortable with the UNIX command line; programmers may find the Python library and API useful as well.
If you use sourmash, please cite us!
Brown and Irber (2016), sourmash: a library for MinHash sketching of DNA Journal of Open Source Software, 1(5), 27, doi:10.21105/joss.00027
sourmash in brief¶
sourmash uses MinHash-style sketching to create “signatures”, compressed representations of DNA/RNA sequence. These signatures can then be stored, searched, explored, and taxonomically annotated.
sourmashprovides command line utilities for creating, comparing, and searching signatures, as well as plotting and clustering signatures by similarity (see the command-line docs).sourmashcan search very large collections of signatures to find matches to a query.sourmashcan also identify parts of metagenomes that match known genomes, and can taxonomically classify genomes and metagenomes against databases of known species.sourmashcan be used to search databases of public sequences (e.g. all of GenBank) and can also be used to create and search databases of private sequencing data.sourmashsupports saving, loading, and communication of signatures via JSON, a ~human-readable & editable format.sourmashalso has a simple Python API for interacting with signatures, including support for online updating and querying of signatures (see the API docs).sourmashisn’t terribly slow, and relies on an underlying Cython module.sourmashis developed on GitHub and is freely and openly available under the BSD 3-clause license. Please see the README for more information on development, support, and contributing.
You can take a look at sourmash analyses on real data in a saved Jupyter notebook, and experiment with it yourself interactively in a Jupyter Notebook at mybinder.org.
Installing sourmash¶
We currently suggest installing the latest pre-release in the sourmash 2.0 series; please see the README file in github.com/dib-lab/sourmash for information. You can use pip or conda equally well.
Memory and speed¶
sourmash has relatively small disk and memory requirements compared to many other software programs used for genome search and taxonomic classification.
First, mash beats sourmash in speed and memory, so if you can use mash,
more power to you :)
sourmash search and sourmash gather can be used to search all
genbank microbial genomes (using our prepared
databases) with about 20 GB of disk and in under 1 GB
of RAM. Typically a search for a single genome takes about 30 seconds
on a laptop.
sourmash lca can be used to search/classify against all genbank
microbial genomes with about 200 MB of disk space and about 10 GB of
RAM. Typically a metagenome classification takes about 1 minute on a
laptop.
Limitations¶
sourmash cannot find matches across large evolutionary distances.
sourmash seems to work well to search and compare data sets for matches at the species and genus level, but does not have much sensitivity beyond that. (It seems to be particularly good at strain-level analysis.) You should use protein-based analyses to do searches across larger evolutionary distances.
sourmash signatures can be very large.
We use a modification of the MinHash sketch approach that allows us to search the contents of metagenomes and large genomes with no loss of sensitivity, but there is a tradeoff: there is no guaranteed limit to signature size when using ‘scaled’ signatures.
Contents:¶
Using sourmash from the command line¶
From the command line, sourmash can be used to compute MinHash sketches from DNA sequences, compare them to each other, and plot the results; these sketches are saved into “signature files”. These signatures allow you to estimate sequence similarity quickly and accurately in large collections, among other capabilities.
Please see the mash software and the mash paper (Ondov et al., 2016) for background information on how and why MinHash sketches work.
sourmash uses a subcommand syntax, so all commands start with
sourmash followed by a subcommand specifying the action to be
taken.
An example¶
Grab three bacterial genomes from NCBI:
curl -L -O ftp://ftp.ncbi.nlm.nih.gov/genomes/refseq/bacteria/Escherichia_coli/reference/GCF_000005845.2_ASM584v2/GCF_000005845.2_ASM584v2_genomic.fna.gz
curl -L -O ftp://ftp.ncbi.nlm.nih.gov/genomes/refseq/bacteria/Salmonella_enterica/reference/GCF_000006945.2_ASM694v2/GCF_000006945.2_ASM694v2_genomic.fna.gz
curl -L -O ftp://ftp.ncbi.nlm.nih.gov/genomes/refseq/bacteria/Sphingobacteriaceae_bacterium_DW12/latest_assembly_versions/GCF_000783305.1_ASM78330v1/GCF_000783305.1_ASM78330v1_genomic.fna.gz
Compute signatures for each:
sourmash compute -k 31 *.fna.gz
This will produce three .sig files containing MinHash signatures at k=31.
Next, compare all the signatures to each other:
sourmash compare *.sig -o cmp
Optionally, parallelize compare to 8 threads with -p 8:
sourmash compare -p 8 *.sig -o cmp
Finally, plot a dendrogram:
sourmash plot cmp --labels
This will output two files, cmp.dendro.png and cmp.matrix.png,
containing a clustering & dendrogram of the sequences, as well as a
similarity matrix and heatmap.
Matrix:
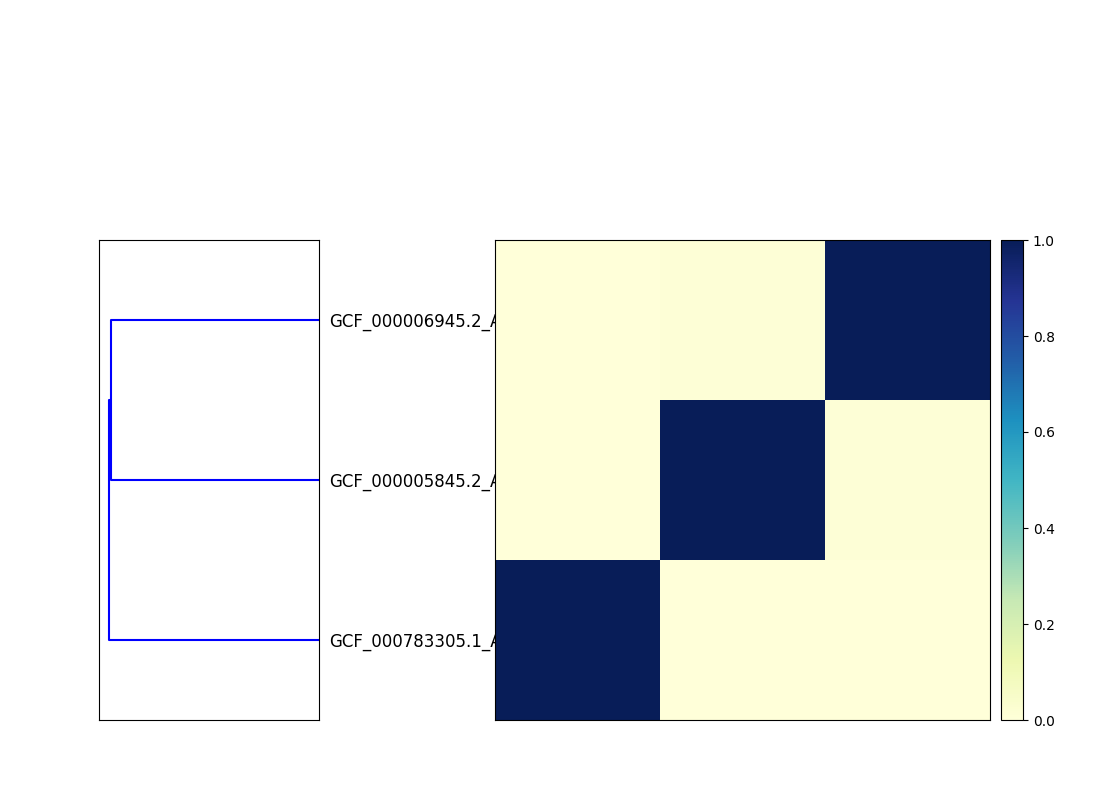 Matrix
Matrix
The sourmash command and its subcommands¶
To get a list of subcommands, run sourmash without any arguments.
There are five main subcommands: compute, compare, plot,
search, and gather. See the tutorial for a
walkthrough of these commands.
computecreates signatures.comparecompares signatures and builds a distance matrix.plotplots distance matrices created bycompare.searchfinds matches to a query signature in a collection of signatures.gatherfinds non-overlapping matches to a metagenome in a collection of signatures.
There are also a number of commands that work with taxonomic
information; these are grouped under the sourmash lca
subcommand. See the LCA tutorial for a
walkthrough of these commands.
lca classifyclassifies many signatures against an LCA database.lca summarizesummarizes the content of a metagenome using an LCA database.lca gatherfinds non-overlapping matches to a metagenome in an LCA database.lca indexcreates a database for use with LCA subcommands.lca rankinfosummarizes the content of a database.lca compare_csvcompares lineage spreadsheets, e.g. those output bylca classify.
Finally, there are a number of utility and information commands:
infoshows version and software information.indexindexes many signatures using a Sequence Bloom Tree (SBT).sbt_combinecombines multiple SBTs.categorizeis an experimental command to categorize many signatures.watchis an experimental command to classify a stream of sequencing data.
sourmash compute¶
The compute subcommand computes and saves signatures for
each sequence in one or more sequence files. It takes as input FASTA
or FASTQ files, and these files can be uncompressed or compressed with
gzip or bzip2. The output will be one or more JSON signature files
that can be used with sourmash compare.
Please see Using sourmash: a practical guide for more information on computing signatures.
Usage:
sourmash compute filename [ filename2 ... ]
Optional arguments:
--ksizes K1[,K2,K3] -- one or more k-mer sizes to use; default is 31
--force -- recompute existing signatures; convert non-DNA characters to N
--output -- save all the signatures to this file; can be '-' for stdout.
--track-abundance -- compute and save k-mer abundances.
--name-from-first -- name the signature based on the first sequence in the file
--singleton -- instead of computing a single signature for each input file,
compute one for each sequence
--merged <name> -- compute a single signature for all of the input files,
naming it <name>
sourmash compare¶
The compare subcommand compares one or more signature files
(created with compute) using estimated Jaccard index.
The default output
is a text display of a similarity matrix where each entry [i, j]
contains the estimated Jaccard index between input signature i and
input signature j. The output matrix can be saved to a file
with --output and used with the sourmash plot subcommand (or loaded
with numpy.load(...). Using --csv will output a CSV file that can
be loaded into other languages than Python, such as R.
Usage:
sourmash compare file1.sig [ file2.sig ... ]
Options:
--output -- save the distance matrix to this file (as a numpy binary matrix)
--ksize -- do the comparisons at this k-mer size.
sourmash plot¶
The plot subcommand produces two plots – a dendrogram and a
dendrogram+matrix – from a distance matrix computed by sourmash compare --output <matrix>. The default output is two PNG files.
Usage:
sourmash plot <matrix>
Options:
--pdf -- output PDF files.
--labels -- display the signature names (by default, the filenames) on the plot
--indices -- turn off index display on the plot.
--vmax -- maximum value (default 1.0) for heatmap.
--vmin -- minimum value (default 0.0) for heatmap.
--subsample=<N> -- plot a maximum of <N> samples, randomly chosen.
--subsample-seed=<seed> -- seed for pseudorandom number generator.
Example output:
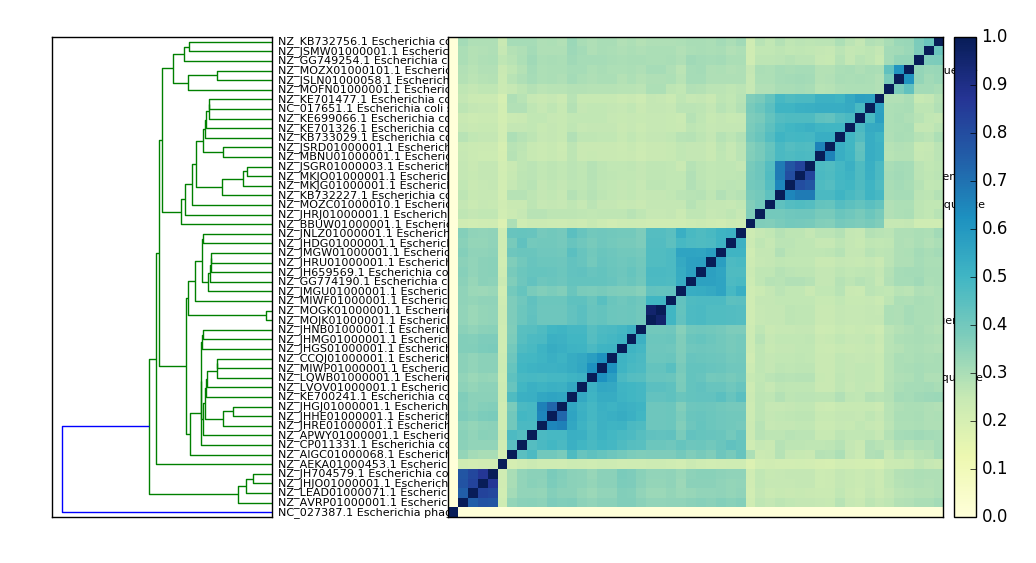 An E. coli comparison plot
An E. coli comparison plot
sourmash search¶
The search subcommand searches a collection of signatures or SBTs for
matches to the query signature. It can search for matches with either
high Jaccard similarity
or containment; the default is to use Jaccard similarity, unless
--containment is specified. -o/--output will create a CSV file
containing the matches.
search will load all of provided signatures into memory, which can
be slow and somewhat memory intensive for large collections. You can
use sourmash index to create a Sequence Bloom Tree (SBT) that can
be quickly searched on disk; this is the same format in which we provide
GenBank and other databases.
Usage:
sourmash search query.sig [ list of signatures or SBTs ]
Example output:
49 matches; showing first 20:
similarity match
---------- -----
75.4% NZ_JMGW01000001.1 Escherichia coli 1-176-05_S4_C2 e117605...
72.2% NZ_GG774190.1 Escherichia coli MS 196-1 Scfld2538, whole ...
71.4% NZ_JMGU01000001.1 Escherichia coli 2-011-08_S3_C2 e201108...
70.1% NZ_JHRU01000001.1 Escherichia coli strain 100854 100854_1...
69.0% NZ_JH659569.1 Escherichia coli M919 supercont2.1, whole g...
...
sourmash gather¶
The gather subcommand finds all non-overlapping matches to the
query. This is specifically meant for metagenome and genome bin
analysis. (See Classifying Signatures
for more information on the different approaches that can be used
here.)
If the input signature was computed with --track-abundance, output
will be abundance weighted (unless --ignore-abundances is
specified). -o/--output will create a CSV file containing the
matches.
gather, like search, will load all of provided signatures into
memory. You can use sourmash index to create a Sequence Bloom Tree
(SBT) that can be quickly searched on disk; this is
the same format in which we provide GenBank and other databases.
Usage:
sourmash gather query.sig [ list of signatures or SBTs ]
Example output:
overlap p_query p_match
--------- ------- --------
1.4 Mbp 11.0% 58.0% JANA01000001.1 Fusobacterium sp. OBRC...
1.0 Mbp 7.7% 25.9% CP001957.1 Haloferax volcanii DS2 pla...
0.9 Mbp 7.4% 11.8% BA000019.2 Nostoc sp. PCC 7120 DNA, c...
0.7 Mbp 5.9% 23.0% FOVK01000036.1 Proteiniclasticum rumi...
0.7 Mbp 5.3% 17.6% AE017285.1 Desulfovibrio vulgaris sub...
Note:
Use sourmash gather to classify a metagenome against a collection of
genomes with no (or incomplete) taxonomic information. Use sourmash lca summarize and sourmash lca gather to classify a metagenome
using a collection of genomes with taxonomic information.
sourmash lca subcommands for taxonomic classification¶
These commands use LCA databases (created with lca index, below, or
prepared databases such as
genbank-k31.lca.json.gz).
sourmash lca classify¶
sourmash lca classify classifies one or more signatures using the given
list of LCA DBs. It is meant for classifying metagenome-assembled genome
bins (MAGs) and single-cell genomes (SAGs).
Usage:
sourmash lca classify --query query.sig [query2.sig ...] --db <lca db> [<lca db2> ...]
For example, the command
sourmash lca classify --query tests/test-data/63.fa.sig \
--db podar-ref.lca.json
will produce the following logging to stderr:
loaded 1 LCA databases. ksize=31, scaled=10000
finding query signatures...
outputting classifications to stdout
... classifying NC_011663.1 Shewanella baltica OS223, complete genome
classified 1 signatures total
and the example classification output is a CSV file with headers:
ID,status,superkingdom,phylum,class,order,family,genus,species
"NC_009665.1 Shewanella baltica OS185, complete genome",found,Bacteria,Proteobacteria,Gammaproteobacteria,Alteromonadales,Shewanellaceae,Shewanella,Shewanella baltica
The status column in the classification output can take three
possible values: nomatch, found, and disagree. nomatch means
that no match was found for this query, and found means that an
unambiguous assignment was found - all k-mers were classified within
the same taxonomic hierarchy, and the most detailed lineage available
was reported. disagree means that there was a taxonomic disagreement,
and the lowest compatible taxonomic node was reported.
To elaborate on this a bit, suppose that all of the k-mers within a
signature were classified as family Shewanellaceae, genus
Shewanella, or species Shewanella baltica. Then the lowest
compatible node (here species Shewanella baltica) would be reported,
and the status of the classification would be found. However, if a
number of additional k-mers in the input signature were classified as
Shewanella oneidensis, sourmash would be unable to resolve the
taxonomic assignment below genus Shewanella and it would report
a status of disagree with the genus-level assignment of Shewanella;
species level assignments would not be reported.
(This is the approach that Kraken and other lowest common ancestor implementations use, we believe.)
sourmash lca summarize¶
sourmash lca summarize produces a Kraken-style summary of the
combined contents of the given query signatures. It is meant for
exploring metagenomes and metagenome-assembled genome bins.
Note, unlike sourmash lca classify, lca summarize merges all
of the query signatures into one and reports on the combined contents.
This may be changed in the future.
Usage:
sourmash lca summarize --query query.sig [query2.sig ...]
--db <lca db> [<lca db2> ...]
For example, the command line:
sourmash lca summarize --query tests/test-data/63.fa.sig \
--db tests/test-data/podar-ref.lca.json
will produce the following log output to stderr:
loaded 1 LCA databases. ksize=31, scaled=10000
finding query signatures...
loaded 1 signatures from 1 files total.
and the following example summarize output to stdout:
50.5% 278 Bacteria;Proteobacteria;Gammaproteobacteria;Alteromonadales;Shewanellaceae;Shewanella;Shewanella baltica;Shewanella baltica OS223
100.0% 550 Bacteria;Proteobacteria;Gammaproteobacteria;Alteromonadales;Shewanellaceae;Shewanella;Shewanella baltica
100.0% 550 Bacteria;Proteobacteria;Gammaproteobacteria;Alteromonadales;Shewanellaceae;Shewanella
100.0% 550 Bacteria;Proteobacteria;Gammaproteobacteria;Alteromonadales;Shewanellaceae
100.0% 550 Bacteria;Proteobacteria;Gammaproteobacteria;Alteromonadales
100.0% 550 Bacteria;Proteobacteria;Gammaproteobacteria
100.0% 550 Bacteria;Proteobacteria
100.0% 550 Bacteria
The output is space-separated and consists of three columns: the percentage of total k-mers that have this classification; the number of k-mers that have this classification; and the lineage classification. K-mer classifications are reported hierarchically, so the percentages and totals contain all assignments that are at a lower taxonomic level - e.g. Bacteria, above, contains all the k-mers in Bacteria;Proteobacteria.
The same information is reported in a CSV file if -o/--output is used.
sourmash lca gather¶
The sourmash lca gather command finds all non-overlapping
matches to the query, similar to the sourmash gather command. This
is specifically meant for metagenome and genome bin analysis. (See
Classifying Signatures for more
information on the different approaches that can be used here.)
If the input signature was computed with --track-abundance, output
will be abundance weighted (unless --ignore-abundances is
specified). -o/--output will create a CSV file containing the
matches.
Usage:
sourmash lca gather query.sig [<lca database> ...]
Example output:
overlap p_query p_match
--------- ------- --------
1.8 Mbp 14.6% 9.1% Fusobacterium nucleatum
1.0 Mbp 7.8% 16.3% Proteiniclasticum ruminis
1.0 Mbp 7.7% 25.9% Haloferax volcanii
0.9 Mbp 7.4% 11.8% Nostoc sp. PCC 7120
0.9 Mbp 7.0% 5.8% Shewanella baltica
0.8 Mbp 6.0% 8.6% Desulfovibrio vulgaris
0.6 Mbp 4.9% 12.6% Thermus thermophilus
sourmash lca index¶
The sourmash lca index command creates an LCA database from
a lineage spreadsheet and a collection of signatures. This can be used
to create LCA databases from private collections of genomes, and can
also be used to create databases for e.g. subsets of GenBank.
See the sourmash lca tutorial and the blog
post
Why are taxonomic assignments so different for Tara bins?
for some use cases.
If you are interested in preparing lineage spreadsheets from GenBank genomes (or building off of NCBI taxonomies more generally), please see the NCBI lineage repository.
sourmash lca rankinfo¶
The sourmash lca rankinfo command displays k-mer specificity
information for one or more LCA databases. See the blog post
How specific are k-mers for taxonomic assignment of microbes, anyway? for example output.
sourmash lca compare_csv¶
The sourmash lca compare_csv command compares two lineage
spreadsheets (such as those output by sourmash lca classify or taken
as input by sourmash lca index) and summarizes their
agreement/disagreement. Please see the blog post
Why are taxonomic assignments so different for Tara bins?
for an example use case.
sourmash signature subcommands for signature manipulation¶
These commands manipulate signatures from the command line. Currently
supported subcommands are merge, rename, intersect,
extract, downsample, subtract, import, export, info,
flatten, and filter.
The signature commands that combine or otherwise have multiple
signatures interacting (merge, intersect, subtract) work only on
compatible signatures, where the k-mer size and nucleotide/protein
sequences match each other. If working directly with the hash values
(e.g. merge, intersect, subtract) then the scaled values must
also match; you can use downsample to convert a bunch of samples to
the same scaled value.
If there are multiple signatures in a file with different ksizes and/or
from nucleotide and protein sequences, you can choose amongst them with
-k/--ksize and --dna or --protein, as with other sourmash commands
such as search, gather, and compare.
Note, you can use sourmash sig as shorthand for all of these commands.
sourmash signature merge¶
Merge two (or more) signatures.
For example,
sourmash signature merge file1.sig file2.sig -o merged.sig
will output the union of all the hashes in file1.sig and file2.sig
to merged.sig.
All of the signatures passed to merge must either have been computed
with --track-abundance, or not. If they have track_abundance on,
then the merged signature will have the sum of all abundances across
the individual signatures. The --flatten flag will override this
behavior and allow merging of mixtures by removing all abundances.
sourmash signature rename¶
Rename the display name for one or more signatures - this is the name
output for matches in compare, search, gather, etc.
For example,
sourmash signature rename file1.sig "new name" -o renamed.sig
will place a renamed copy of the hashes in file1.sig in the file
renamed.sig. If you provide multiple signatures, all will be renamed
to the same name.
sourmash signature subtract¶
Subtract all of the hash values from one signature that are in one or more of the others.
For example,
sourmash signature subtract file1.sig file2.sig file3.sig -o subtracted.sig
will subtract all of the hashes in file2.sig and file3.sig from
file1.sig, and save the new signature to subtracted.sig.
To use subtract on signatures calculated with
--track-abundance, you must specify --flatten.
sourmash signature intersect¶
Output the intersection of the hash values in multiple signature files.
For example,
sourmash signature intersect file1.sig file2.sig file3.sig -o intersect.sig
will output the intersection of all the hashes in those three files to
intersect.sig.
The intersect command flattens all signatures, i.e. the abundances
in any signatures will be ignored and the output signature will have
track_abundance turned off.
sourmash signature downsample¶
Downsample one or more signatures.
With downsample, you can –
- increase the
--scaledvalue for a signature computed with--scaled, shrinking it in size; - decrease the
numvalue for a traditional num MinHash, shrinking it in size; - try to convert a
--scaledsignature to anumsignature; - try to convert a
numsignature to a--scaledsignature.
For example,
sourmash signature downsample file1.sig file2.sig --scaled 100000 -o downsampled.sig
will output each signature, downsampled to a scaled value of 100000, to
downsampled.sig; and
sourmash signature downsample --num 500 scaled_file.sig -o downsampled.sig
will try to convert a scaled MinHash to a num MinHash.
sourmash signature extract¶
Extract the specified signature(s) from a collection of signatures.
For example,
sourmash signature extract *.sig -k 21 --dna -o extracted.sig
will extract all nucleotide signatures calculated at k=21 from all .sig files in the current directory.
There are currently two other useful selectors for extract: you can specify
(part of) an md5sum, as output in the CSVs produced by search and gather;
and you can specify (part of) a name.
For example,
sourmash signature extract tests/test-data/*.fa.sig --md5 09a0869
will extract the signature from 47.fa.sig which has an md5sum of
09a08691ce52952152f0e866a59f6261; and
sourmash signature extract tests/test-data/*.fa.sig --name NC_009665
will extract the same signature, which has an accession number of
NC_009665.1.
sourmash signature flatten¶
Flatten the specified signature(s), removing abundances and setting track_abundance to False.
For example,
sourmash signature flatten *.sig -o flattened.sig
will remove all abundances from all of the .sig files in the current directory.
The flatten command accepts the same selectors as extract.
sourmash signature filter¶
Filter the hashes in the specified signature(s) by abundance, by either
-m/--min-abundance or -M/--max-abundance or both. Abundance selection is
inclusive, so -m 2 -M 5 will select hashes with abundance greater than
or equal to 2, and less than or equal to 5.
For example,
sourmash signature -m 2 *.sig
will output new signatures containing only hashes that occur two or more times in each signature.
The filter command accepts the same selectors as extract.
sourmash signature import¶
Import signatures into sourmash format. Currently only supports mash,
and can import mash sketches output by mash info -d <filename.msh>.
For example,
sourmash signature import filename.msh.json -o imported.sig
will import the contents of filename.msh.json into imported.sig.
sourmash signature export¶
Export signatures from sourmash format. Currently only supports mash dump format.
For example,
sourmash signature export filename.sig -o filename.sig.msh.json
sourmash signature overlap¶
Display a detailed comparison of two signatures. This computes the
Jaccard similarity (as in sourmash compare or sourmash search) and
the Jaccard containment in both directions (as with --containment).
It also displays the number of hash values in the union and
intersection of the two signatures, as well as the number of disjoint
hash values in each signature.
This command has two uses - first, it is helpful for understanding how similarity and containment are calculated, and second, it is useful for analyzing signatures with very small overlaps, where the similarity and/or containment might be very close to zero.
For example,
sourmash signature overlap file1.sig file2.sig
will display the detailed comparison of file1.sig and file2.sig.
sourmash tutorials and notebooks¶
The first two tutorials!¶
These tutorials are both command line tutorials that should work on Mac OS X and Linux. They require about 5 GB of disk space and 5 GB of RAM.
Background and details¶
These next three tutorials are all notebooks that you can view, run yourself, or run interactively online via the binder service.
More information¶
If you are a Python programmer, you might also be interested in our API examples.
If you prefer R, we have a short guide to using sourmash output with R.
Using sourmash: a practical guide¶
So! You’ve installed sourmash, run a few of the tutorials and commands, and now you actually want to use it. This guide is here to answer some of your questions, and explain why we can’t answer others.
(If you have additional questions, please file an issue!)
What k-mer size(s) should I use?¶
You can build signatures at a variety of k-mer sizes all at once, and (unless you are working with very large metagenomes) the resulting signature files will still be quite small.
We suggest including k=31 and k=51. k=51 gives you the most stringent matches, and has very few false positives. k=31 may be more sensitive at the genus level.
Why 31 and 51, specifically? To a large extent these numbers were picked out of a hat, based on our reading of papers like the Metapalette paper (Koslicki and Falush, 2016. You could go with k=49 or k=53 and probably get very similar results to k=51. The general rule is that longer k-mer sizes are less prone to false positives. But you can pick your own parameters.
One additional wrinkle is that we provide a number of precomputed databases at k=21, k=31, and k=51. It is often convenient to calculate signatures at these sizes so that you can use these databases.
You’ll notice that all of the above numbers are odd. That is to avoid occasional minor complications from palindromes in numerical calculations, where the forward and reverse complements of a k-mer are identical. This cannot happen if k is odd. It is not enforced by sourmash, however, and it probably doesn’t really matter.
(When we have blog posts or publications providing more formal guidance, we’ll link to them here!)
What resolution should my signatures be / how should I compute them?¶
sourmash supports two ways of choosing the resolution or size of
your signatures: using -n to specify the maximum number of hashes,
or --scaled to specify the compression ratio. Which should you use?
We suggest calculating all your signatures using --scaled 1000. This will give you a compression ratio of 1000-to-1 while
making it possible to detect regions of similarity in the 10kb range.
For comparison with more traditional MinHash approaches like mash,
if you have a 5 Mbp genome and use --scaled 1000, you will extract
approximately 5000 hashes. So a scaled of 1000 is equivalent to using
-n 5000 with mash on a 5 Mbp genome.
The difference between using -n and --scaled is in metagenome
analysis: fixing the number of hashes with -n limits your ability to
detect rare organisms, or alternatively results in very large
signatures (e.g. if you use n larger than 10000). --scaled will scale
your resolution with the diversity of the metagenome.
You can read more about this in this blog post from the mash folk,
Mash Screen: What’s in my sequencing run? What
we do with sourmash and --scaled is similar to the ‘modulo hash’
mentioned in that blog post.
(Again, when we have formal guidance on this based on benchmarks, we’ll link to it here.)
What kind of input data does sourmash work on?¶
sourmash has been used most extensively with Illumina read data sets and assembled genomes, transcriptomes, and metagenomes. The high error rate of PacBio and Nanopore sequencing is problematic for k-mer based approaches and we have not yet explored how to tune parameters for this kind of sequencing.
On a more practical note, sourmash compute should autodetect FASTA,
FASTQ, whether they are uncompressed, gzipped, or bzip2-ed. Nothing
special needs to be done.
How should I prepare my data?¶
Raw Illumina read data sets should be k-mer abundance trimmed to get rid of the bulk of erroneous kmers. We suggest a command like the following, using trim-low-abund from the khmer project –
trim-low-abund.py -C 3 -Z 18 -V -M 2e9 <all of your input read files>
This is safe to use on genomes, metagenomes, and transcriptomes. If you
are working with large genomes or diverse metagenomes, you may need to
increase the -M parameter to use more memory.
See the khmer docs for trim-low-abund.py and the semi-streaming preprint for more information.
For high coverage genomic data, you can do very stringent trimming with an absolute cutoff, e.g.
trim-low-abund.py -C 10 -M 2e9 <all of your input read files>
will eliminate all k-mers that appear fewer than 10 times in your data set. This kind of trimming will dramatically reduce your sensitivity when working with metagenomes and transcriptomes, however, where there are always real low-abundance k-mers present.
Could you just give us the !#%#!$ command line?¶
Sorry, yes! See below.
Computing signatures for read files:¶
trim-low-abund -C 3 -Z 18 -V -M 2e9 input-reads-1.fq input-reads-2.fq ...
sourmash compute --scaled 1000 -k 21,31,51 input-reads*.fq.abundtrim \
--merge SOMENAME -o SOMENAME-reads.sig
The first command trims off low-abundance k-mers from high-coverage
reads; the second takes all the trimmed read files, subsamples k-mers
from them at 1000:1, and outputs a single merged signature named
‘SOMENAME’ into the file SOMENAME-reads.sig.
Computing signatures for individual genome files:¶
sourmash compute --scaled 1000 -k 21,31,51 *.fna.gz --name-from-first
This command computes signatures for all *.fna.gz files, and names
each signature based on the first FASTA header in each file (that’s
what the option --name-from-first does). The signatures will be placed
in *.fna.gz.sig.
Computing signatures from a collection of genomes in a single file:¶
sourmash compute --scaled 1000 -k 21,31,51 file.fa --singleton
This computes signatures for all individual FASTA sequences in file.fa,
names them based on their FASTA headers, and places them all in a single
.sig file, file.fa.sig. (This behavior is triggered by the option
--singleton, which tells sourmash to treat each individual sequence in
the file as an independent sequence.)
Classifying signatures: search, gather, and lca methods.¶
sourmash provides several different techniques for doing classification and breakdown of signatures.
Searching for similar samples with search.¶
The sourmash search command is most useful when you are looking for
high similarity matches to other signatures; this is the most basic use
case for MinHash searching. The command takes a query signature and one
or more search signatures, and finds all the matches it can above a particular
threshold.
By default search will find matches with high Jaccard
similarity, which will
consider all of the k-mers in the union of the two samples.
Practically, this means that you will only find matches if there is
both high overlap between the samples and relatively few k-mers that
are disjoint between the samples. This is effective for finding genomes
or transcriptomes that are similar but rarely works well for samples
of vastly different sizes.
One useful modification to search is to calculate containment with
--containment instead of the (default) similarity; this will find
matches where the query is contained within the subject, but the
subject may have many other k-mers in it. For example, if you are using
a plasmid as a query, you would use --containment to find genomes
that contained that plasmid.
See the main sourmash
tutorial
for information on using search with and without --containment.
Breaking down metagenomic samples with gather and lca¶
Neither search option (similarity or containment) is effective when comparing or searching with metagenomes, which typically have a mixture of many different genomes. While you might use containment to see if a query genome is present in one or more metagenomes, a common question to ask is the reverse: what genomes are in my metagenome?
We have implemented two algorithms in sourmash to do this.
One algorithm uses taxonomic information from e.g. GenBank to classify
individual k-mers, and then infers taxonomic distributions of
metagenome contents from the presence of these individual
k-mers. (This is the approach pioneered by
Kraken and many other tools.)
sourmash lca can be used to classify individual genome bins with
classify, or summarize metagenome taxonomy with summarize. The
sourmash lca tutorial
shows how to use the lca classify and summarize commands, and also
provides guidance on building your own database.
The other approach, gather, breaks a metagenome down into individual
genomes based on greedy partitioning. Essentially, it takes a query
metagenome and searches the database for the most highly contained
genome; it then subtracts that match from the metagenome, and repeats.
At the end it reports how much of the metagenome remains unknown. The
basic sourmash
tutorial
has some sample output from using gather with GenBank.
Our preliminary benchmarking suggests that gather is the most accurate
method available for doing strain-level resolution of genomes. More on that
as we move forward!
To do taxonomy, or not to do taxonomy?¶
By default, there is no structured taxonomic information available in sourmash signatures or SBT databases of signatures. Generally what this means is that you will have to provide your own mapping from a match to some taxonomic hierarchy. This is generally the case when you are working with lots of genomes that have no taxonomic information.
The lca subcommands, however, work with LCA databases, which contain
taxonomic information by construction. This is one of the main
differences between the sourmash lca subcommands and the basic
sourmash search functionality. So the lca subcommands will generally
output structured taxonomic information, and these are what you should look
to if you are interested in doing classification.
The command lca gather applies the gather algorithm to search an
LCA database; it reports taxonomy.
It’s important to note that taxonomy based on k-mers is very, very specific and if you get a match, it’s pretty reliable. On the converse, however, k-mer identification is very brittle with respect to evolutionary divergence, so if you don’t get a match it may only mean that the particular species isn’t known.
Abundance weighting¶
If you compute your input signatures with --track-abundance, both
sourmash gather and sourmash lca gather will use that information
to calculate an abundance-weighted result. Briefly, this will weight
each match to a hash value by the multiplicity of the hash value in
the query signature. You can turn off this behavior with
--ignore-abundance.
What commands should I use?¶
It’s not always easy to figure that out, we know! We’re thinking about better tutorials and documentation constantly.
We suggest the following approach:
- build some signatures and do some searches, to get some basic familiarity with sourmash;
- explore the available databases;
- then ask questions via the issue tracker and we will do our best to help you out!
This helps us figure out what people are actually interested in doing, and any help we provide via the issue tracker will eventually be added into the documentation.
sourmash Python API¶
The primary programmatic way of interacting with sourmash is via
its Python API. Please also see examples of using the API.
Contents
MinHash: basic MinHash sketch functionality¶
An implementation of a MinHash bottom sketch, applied to k-mers in DNA.
SourmashSignature: save and load MinHash sketches in JSON¶
Save and load MinHash sketches in a JSON format, along with some metadata.
-
class
sourmash.signature.SourmashSignature(minhash, name='', filename='')[source]¶ Main class for signature information.
SBT: save and load Sequence Bloom Trees in JSON¶
An implementation of sequence bloom trees, Solomon & Kingsford, 2015.
To try it out, do:
factory = GraphFactory(ksize, tablesizes, n_tables)
root = Node(factory)
graph1 = factory()
# ... add stuff to graph1 ...
leaf1 = Leaf("a", graph1)
root.add_node(leaf1)
For example,
# filenames: list of fa/fq files
# ksize: k-mer size
# tablesizes: Bloom filter table sizes
# n_tables: Number of tables
factory = GraphFactory(ksize, tablesizes, n_tables)
root = Node(factory)
for filename in filenames:
graph = factory()
graph.consume_fasta(filename)
leaf = Leaf(filename, graph)
root.add_node(leaf)
then define a search function,
def kmers(k, seq):
for start in range(len(seq) - k + 1):
yield seq[start:start + k]
def search_transcript(node, seq, threshold):
presence = [ node.data.get(kmer) for kmer in kmers(ksize, seq) ]
if sum(presence) >= int(threshold * len(seq)):
return 1
return 0
-
class
sourmash.sbt.GraphFactory(ksize, starting_size, n_tables)[source]¶ Build new nodegraphs (Bloom filters) of a specific (fixed) size.
Parameters: - ksize (int) – k-mer size.
- starting_size (int) – size (in bytes) for each nodegraph table.
- n_tables (int) – number of nodegraph tables to be used.
-
class
sourmash.sbt.Node(factory, name=None, path=None, storage=None)[source]¶ Internal node of SBT.
-
data¶
-
-
class
sourmash.sbt.SBT(factory, d=2, storage=None)[source]¶ A Sequence Bloom Tree implementation allowing generic internal nodes and leaves.
The default node and leaf format is a Bloom Filter (like the original implementation), but we also provide a MinHash leaf class (in the sourmash.sbtmh.SigLeaf class)
Parameters: - factory (Factory) – Callable for generating new datastores for internal nodes.
- d (int) – Number of children for each internal node. Defaults to 2 (a binary tree)
- storage (Storage, default: None) – A Storage is any place where we can save and load data for the nodes. If set to None, will use a FSStorage.
Notes
We use two dicts to store the tree structure: One for the internal nodes, and another for the leaves (datasets).
-
child(parent, pos)[source]¶ Return a child node at position
posunder theparentnode.Parameters: - parent (int) – Parent node position in the tree.
- pos (int) – Position of the child one under the parent. Ranges from [0, arity - 1], where arity is the arity of the SBT (usually it is 2, a binary tree).
Returns: A NodePos namedtuple with the position and content of the child node.
Return type:
-
children(pos)[source]¶ Return all children nodes for node at position
pos.Parameters: pos (int) – Position of the node in the tree. Returns: A list of NodePos namedtuples with the position and content of all children nodes. Return type: list of NodePos
-
classmethod
load(location, leaf_loader=None, storage=None, print_version_warning=True)[source]¶ Load an SBT description from a file.
Parameters: - location (str) – path to the SBT description.
- leaf_loader (function, optional) – function to load leaf nodes. Defaults to
Leaf.load. - storage (Storage, optional) – Storage to be used for saving node data. Defaults to FSStorage (a hidden directory at the same level of path)
Returns: the SBT tree built from the description.
Return type:
-
parent(pos)[source]¶ Return the parent of the node at position
pos.If it is the root node (position 0), returns None.
Parameters: pos (int) – Position of the node in the tree. Returns: A NodePos namedtuple with the position and content of the parent node. Return type: NodePos
-
save(path, storage=None, sparseness=0.0, structure_only=False)[source]¶ Saves an SBT description locally and node data to a storage.
Parameters: - path (str) – path to where the SBT description should be saved.
- storage (Storage, optional) – Storage to be used for saving node data. Defaults to FSStorage (a hidden directory at the same level of path)
- sparseness (float) – How much of the internal nodes should be saved. Defaults to 0.0 (save all internal nodes data), can go up to 1.0 (don’t save any internal nodes data)
- structure_only (boolean) – Write only the index schema and metadata, but not the data. Defaults to False (save data too)
Returns: full path to the new SBT description
Return type: str
sourmash.fig: make plots and figures¶
Make plots using the distance matrix+labels output by sourmash compare.
Additional information on sourmash¶
Computational requirements¶
Read more about the compute requirements, here.
Prepared search database¶
We offer a number of prepared search databases.
Other MinHash implementations for DNA¶
In addition to mash, also see:
- RKMH: Read Classification by Kmers
- mashtree: For building trees using Mash
- Finch: “Fast sketches, count histograms, better filtering.”
- BBMap and SendSketch: part of Brian Bushnell’s tool collection.
- PATRIC uses MinHash for genome search.
If you are interested in exactly how these MinHash approaches calculate the hashes of DNA sequences, please see some simple Python code in sourmash, utils/compute-dna-mh-another-way.py
Papers and references¶
On the resemblance and containment of documents, Broder, 1997. The original MinHash paper!
Mash: fast genome and metagenome distance estimation using MinHash., Ondov et al. 2016.
sourmash: a library for MinHash sketching of DNA., Brown and Irber, 2017.
Improving MinHash via the Containment Index with Applications to Metagenomic Analysis, Koslicki and Zabeti, 2017.
Ultra-fast search of all deposited bacterial and viral genomic data, Bradley et al., 2019.
Streaming histogram sketching for rapid microbiome analytics, Rowe et al., 2019.
Dashing: Fast and Accurate Genomic Distances with HyperLogLog, Baker and Langmead, 2019.
Presentations and posters¶
Taxonomic classification of microbial metagenomes using MinHash signatures, Brooks et al., 2017. Presented at ASM.
Blog posts¶
We (and others) have a number of blog posts on sourmash and MinHash more generally:
- Some background on k-mers, and their use in taxonomy
- From the Phillippy lab: mash screen: what’s in my sequencing run?
- Applying MinHash to cluster RNAseq samples
- MinHash signatures as ways to find samples, and collaborators?
- Efficiently searching MinHash Sketch collections: indexing and search 42,000 bacterial genomes with Sequence Bloom Trees.
- Quickly searching all the microbial genomes, mark 2 - now with archaea, phage, fungi, and protists!: indexing and searching 50,000 microbial genomes, round 2.
- What metadata should we put in MinHash Sketch signatures?: crowdsourcing ideas for what metadata belongs in a signature file.
- Minhashing all the things (part 1): microbial genomes: on approaches to computing MinHashes for large collections of public data.
- Comparing genome sets extracted from metagenomes
- Taxonomic examinations of genome bins from Tara Oceans
- Classifying genome bins using a custom reference database, part I
- Classifying genome bins using a custom reference database, part II
JSON format for the signature¶
The JSON format is not necessarily final; this is a TODO item for future releases. In particular, we’d like to update it to store more metadata for samples.
Interoperability with mash¶
The default sketches computed by sourmash and mash are comparable, but we are still working on ways to convert the file formats
Known issues¶
For at least some versions of matplotlib, users may encounter an error “Failed to connect to server socket:” or “RuntimeError: Invalid DISPLAY variable”. This is because by default matplotlib tries to connect to X11 to use the Tkinter backend.
The solution is to force the use of the ‘Agg’ backend in matplotlib; see this stackoverflow answer or this sourmash issue comment.
Newer versions of matplotlib do not seem to have this problem.
Support¶
Please ask questions and file bug descriptions on the GitHub issuetracker for sourmash, dib-lab/sourmash/issues
You can also ask questions of Titus on Twitter at @ctitusbrown